What is Ketamine?
The fancy name for ketamine is:
2-(2-chlorophenyl)-2-(methylamino)-cyclohexan-1-one
It is an arylcycloalkamine and is structurally related to cyclidines, such as phencyclidine (PCP). It is synthesized using the reagents o-chlorobenzonitrile and cyclopentyl grignard. Ketamine is a racemic mixture of S-(+)-ketamine and R-(-)-ketamine and it acts as a NMDA receptor antagonist. Ketamine modulates the glutamate system, which is different from traditional antidepressant therapies.
Ketamine has been used for both anesthetic purposes and for veterinarian surgical procedures as well.
Ketamine is available as a hydrochloride salt or freebase, and it is water-soluble. It’s availability in the body depends on the route with which it is administered. See the table below:
Route Bioavailability
| Intramuscular | 93% |
| Intranasal | 50% |
| Intravenous | 100% |
| Oral | 20% |
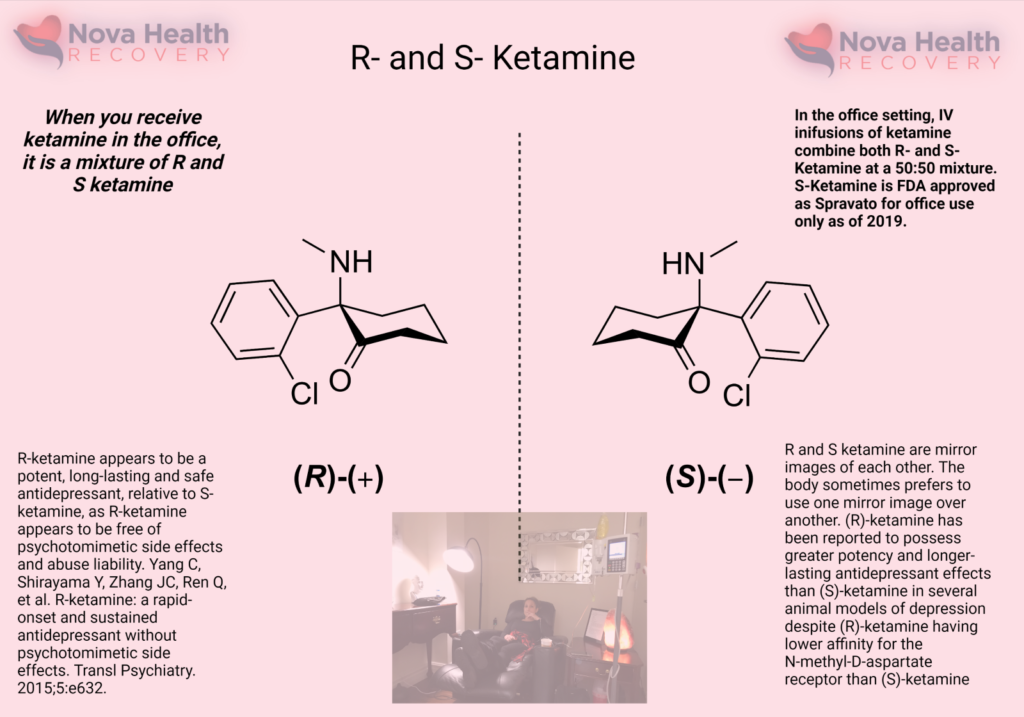
The left-handed form of ketamine or S-ketamine is otherwise known as Spravato, which is an FDA-approved nasal spray of ketamine that has been available since 2019. It is taken alongside an oral antidepressant medication for treatment-resistant depression. Intravenous ketamine is given as a racemic mixture, in which both the right- and left-handed forms of ketamine are administered. The different enantiomers of ketamine, which refers to either the right- or left-handed ketamine, have different pharmacological activity, tissue uptake, metabolism, and receptor affinity.
Pharmacokinetics: Ketamine is highly liposoluble with low protein binding ability, which enables rapid transfer across the blood-brain barrier. Concentrations of ketamine in the brain are often 4-5 times higher than that in the blood plasma. It takes between 7 – 11 minutes for the plasma concentration of ketamine to reduce by 50%.
Ketamine enantiomers:
S-ketamine has a higher affinity for the NMDA receptor and so lower doses are required to achieve effect. (1)(2) As lower doses of S-ketamine are required to elicit effects, there is probably less drowsiness, agitation, and cognitive issues when it is administered. S-ketamine has a 2-3 fold higher affinity for the opioid receptor than R-ketamine.
R-ketamine has a lower NMDA receptor affinity and is not used alone currently. There are studies demonstrating that this enantiomer induces longer antidepressant effects and higher rates of synaptogenesis relative to the S version. (3)(4)(5) The R version also has less dissociation and sensory deficits as well when it is administered. (6)
Ketamine Metabolism:
Ketamine is primarily metabolized via oxidative N-demethylation of the cyclohexanone ring by the CYP450 enzyme, producing the primary and active metabolite, norketamine (80%) which is hydroxylated to hydroxy-norketamine (15%) and conjugated with glucoronate and excreted in the urine.
How does ketamine interact with receptors in the brain?
Ketamine binds the numerous receptors, but its main effect is through its activity as a non-competitive NMDA receptor antagonist, where it blocks the PCP binding site of the NMDA receptor and prevents depolarization of the neuron. Generally speaking, the NMDA receptor is an excitatory receptor playing a role in learning and sensory perception. Ketamine also interacts with other receptors at higher doses, such as the opioid receptors, which may contribute to the anti-pain (antinociceptive) effects of ketamine. (7)(8) Ketamine also interacts with the HCN1 and sigma receptors as an antagonist. It is also an antagonist at muscarinic CNS receptors and agonistic (activating) at alpha- and beta-adrenergic receptors.
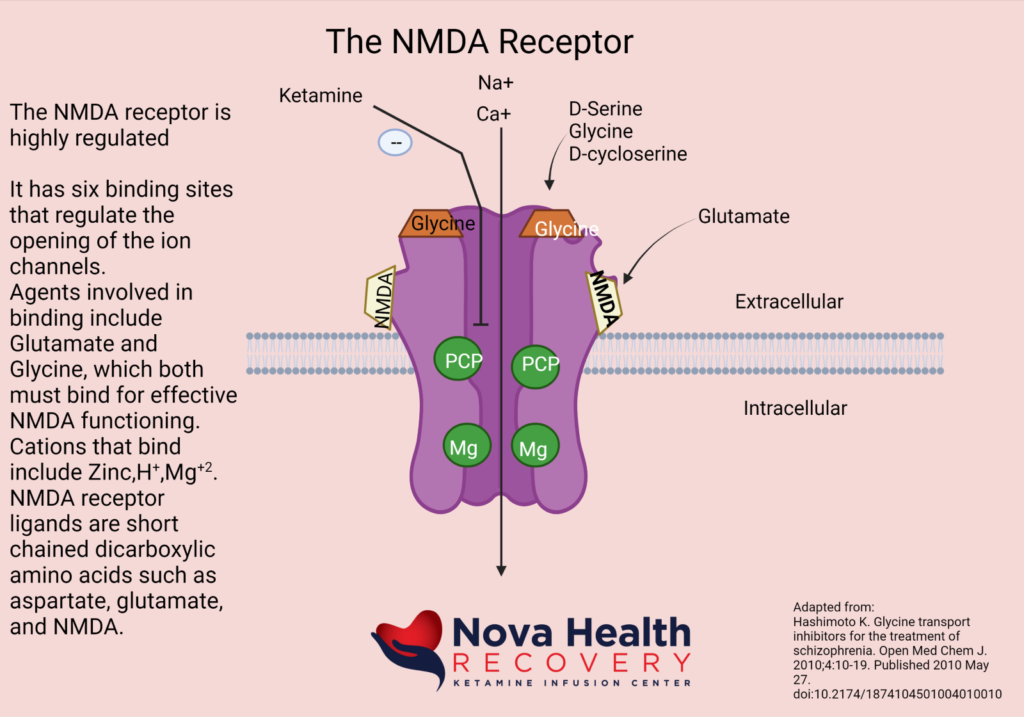
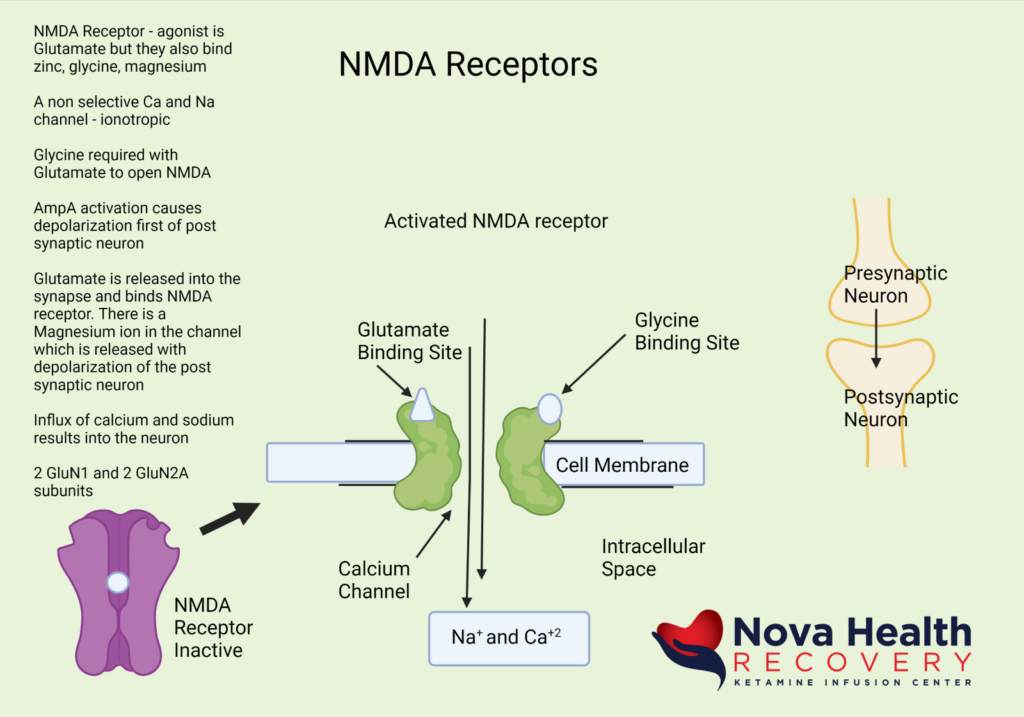
What does the NMDA Receptor look like? How is the NMDA receptor Activated?
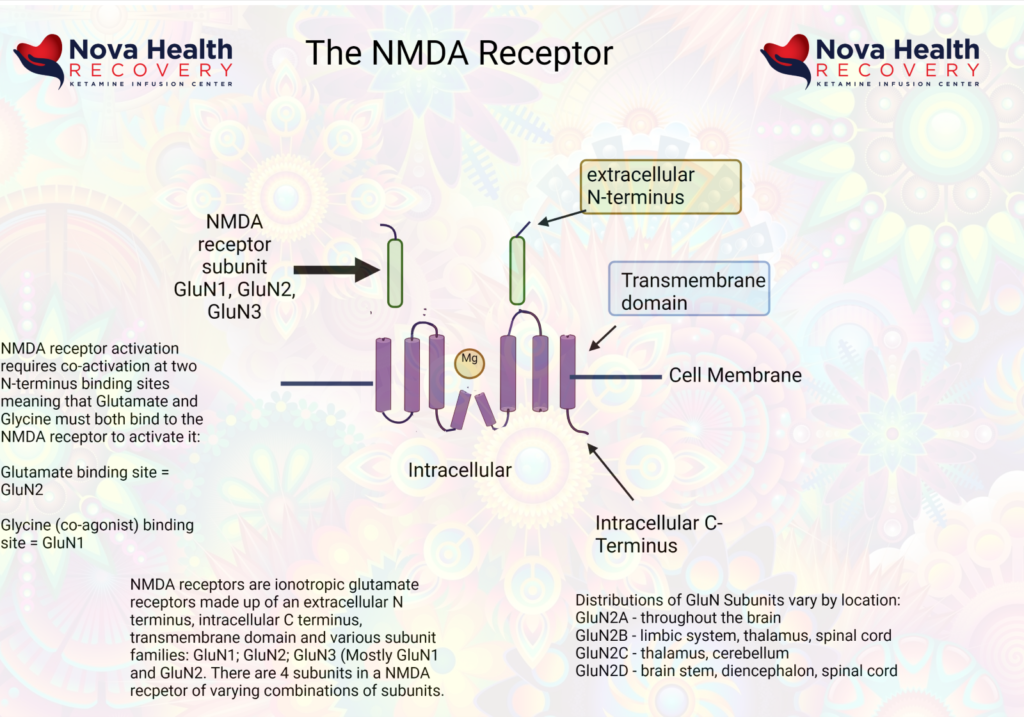
Magnesium blocks the NMDA receptor and prevents inflow of Na+ and Ca+2:
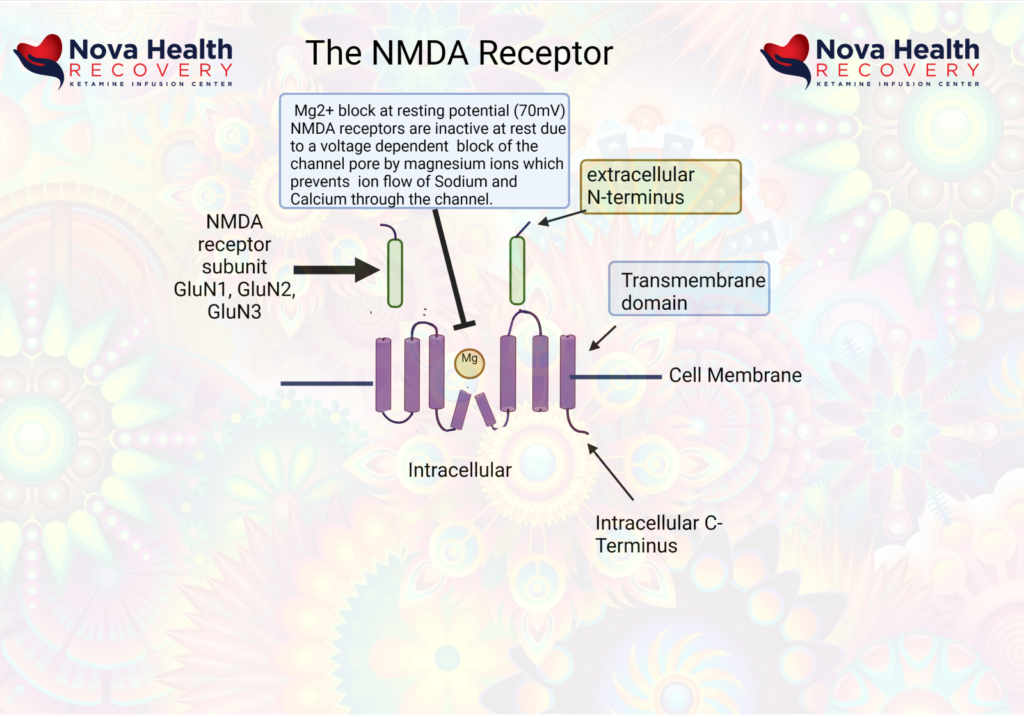
The post-synaptic neuron needs to be depolarized enough to allow the Magnesium ion to be displaced. This first occurs through activation of the AMPA receptor, which also is a receptor that binds/responds to glutamate and is present where there are NMDA receptors.
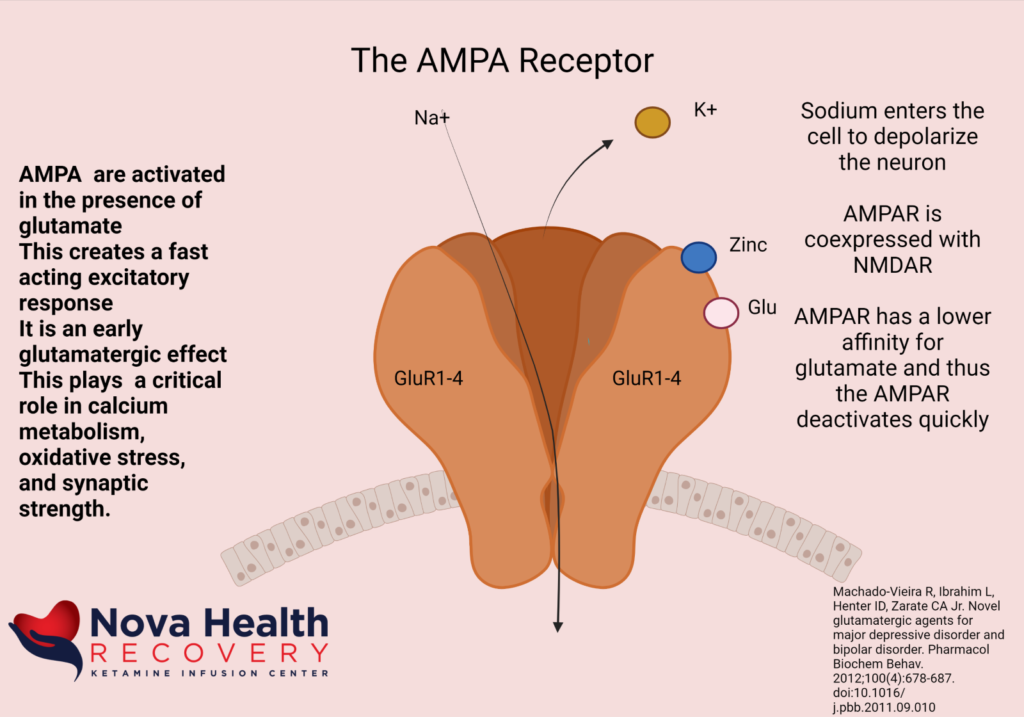
So, the AMPA receptor gets activated first with glutamate binding and allows a current of sodium ions into the post synaptic neuron, which then depolarizes the neuron and pushes out the Magnesium ion in the NMDA channel. The NMDA receptor now can allow sodium and calcium ions into the post-synaptic neuron to cause downstream protein production (BDNF and synaptogenesis).
Again, the sequence of NMDA receptor activation is:
- Low level background glutamate release by pre-synaptic neuron interacts with NMDA and AMPA receptors. The AMPA receptors are more reactive initially and allow sodium into the post-synaptic neuron, which depolarizes it.
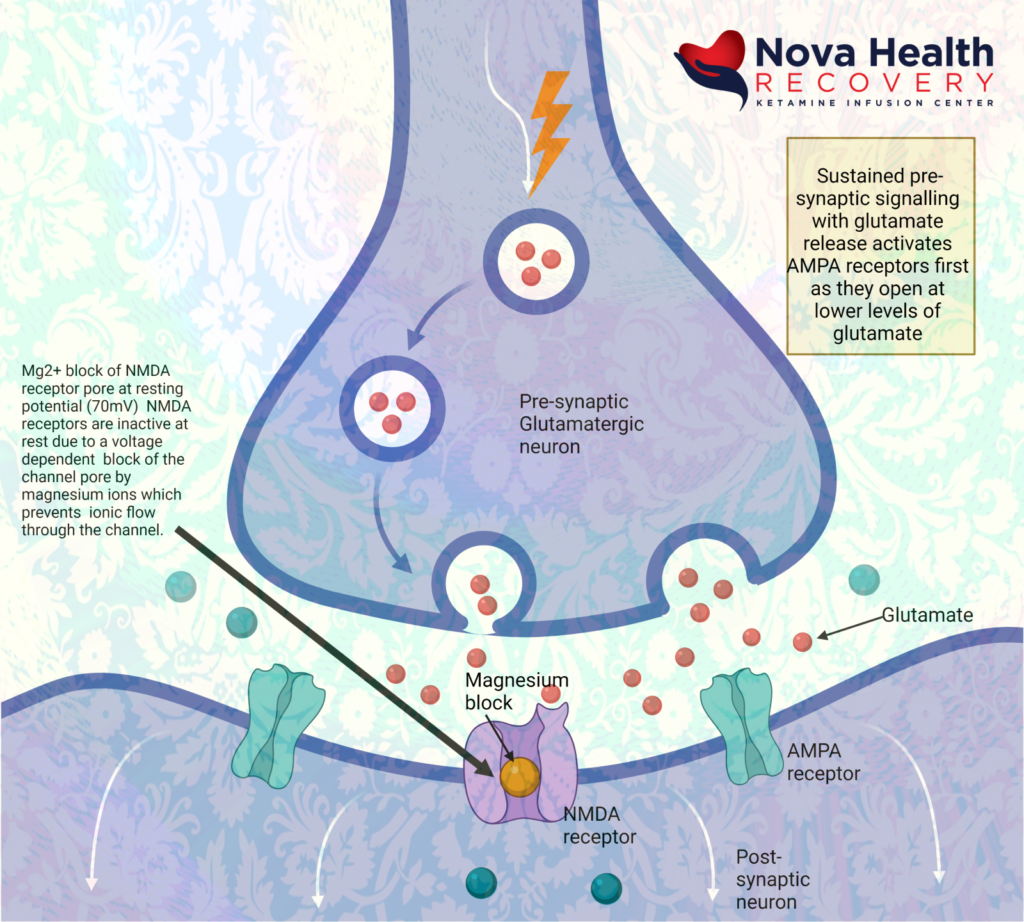
- AMPA receptors open due to binding with glutamate letting in sodium to depolarize post-synaptic neuron:
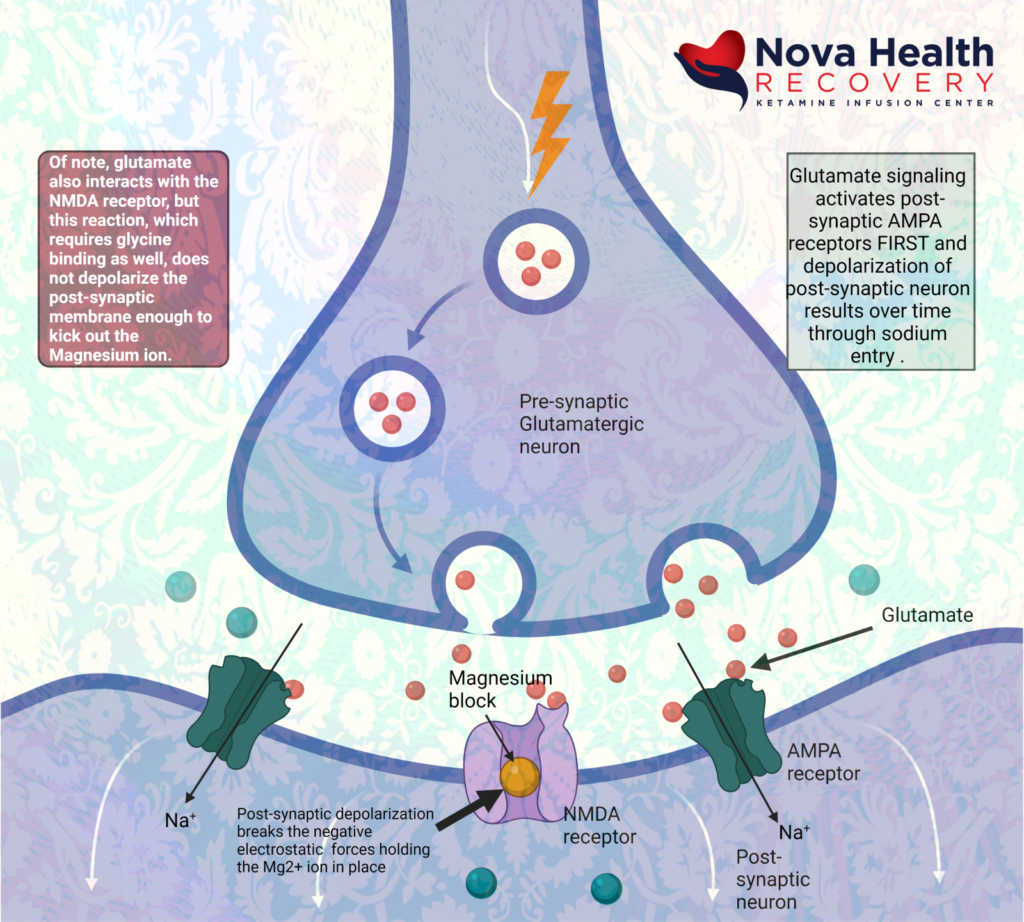
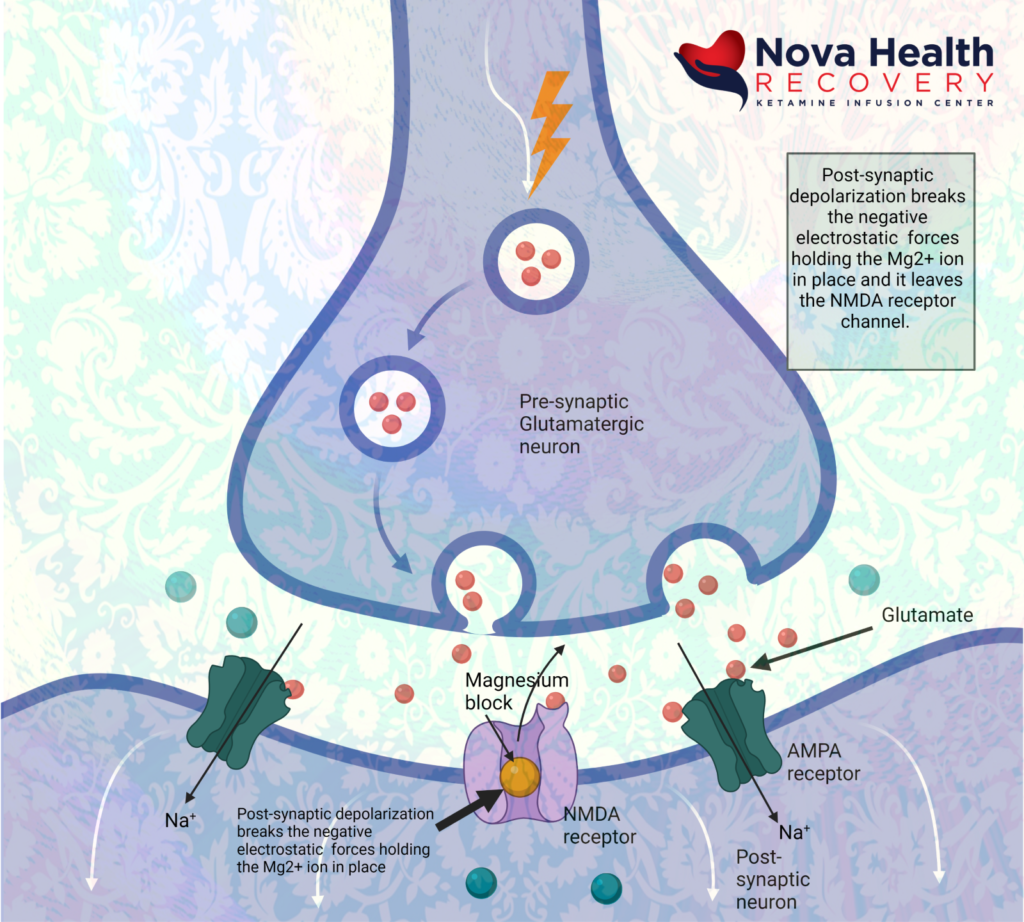
- Magnesium is extruded from the NMDA receptor pore due to depolarization of post-synaptic neuron. Calcium and sodium can enter through the receptor pore now causing metabolic signaling:
Ketamine acts at the internal pore PCP site in the intracellular portion of the pore, where it’s binding on primarily the Glu2N unit results in decreased opening time of the pore and less calcium inflow. This decreases sensory input to higher areas of the CNS, such as emotional response, learning and memory.
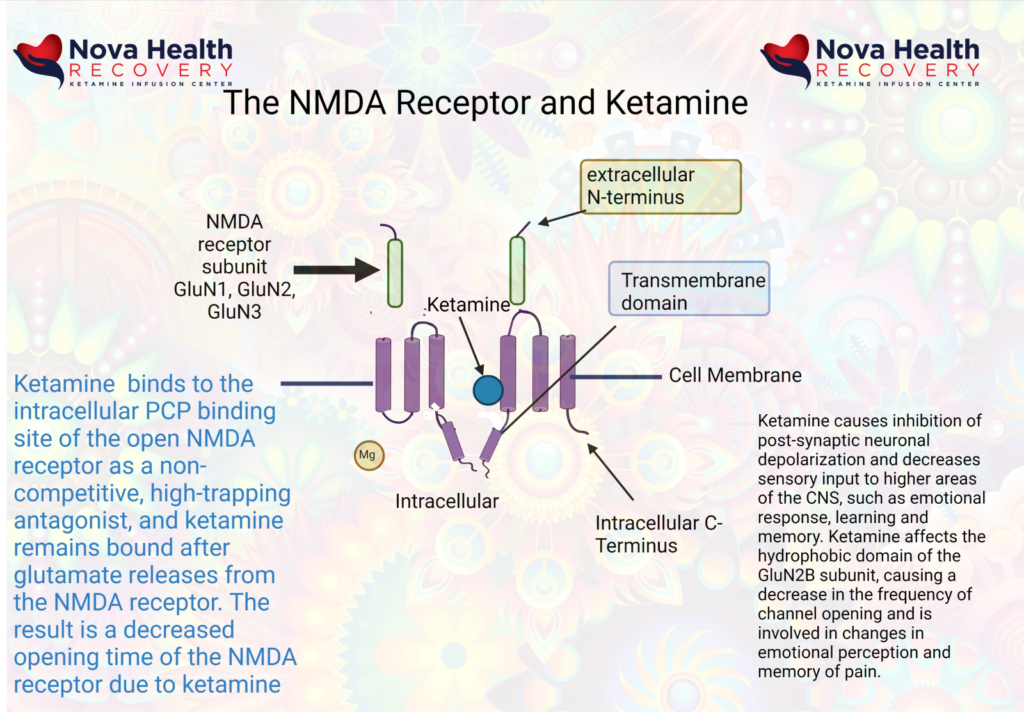
As a result of ketamine’s actions in altering excessive NMDA receptor activation, emotional and pain signaling are modified. Learning and memory may be modified through ketamine’s effects on the hippocampus, and there is enhanced emotional control as the PFC gains control over the limbic brain.
Ketamine has dissociative properties:
Sub-anesthetic doses of ketamine can produce states characterized by hallucinations, feelings of detachment from the environment, delirium, feelings of detachment from self, agitation, visual and auditory distortions all of which are characteristic of a dissociative state. Blood levels of ketamine are generally between 50-100 ng/ml. This state is due to blocking of the NMDA channel by ketamine with dysregulated excitatory neuronal activity. In this state of dissociation, one will find excessive glutamate release, increased dopamine activity, altered synaptic homeostasis, and decreased acetylcholine activity.
Dissociation is a state in which sensory inputs can reach the cerebral cortex but are not perceived in processing areas.
Ketamine and pain management:
Ketamine therapy causes alterations in gene expression and cell signaling that interrupt pathological changes associated with chronic pain. These include:
- Increased concentration of amines (5HT, NA, DA) which increases downward inhibition of pain perception
- Changes the NMDA receptor expression which is altered (by phosphorylation) in chronic pain states
- Ketamine acts as an anti-inflammatory agent and can downregulate activated astrocytes that may be producing inflammatory changes present in chronic pain
- Increased synaptic plasticity (neuroplasticity)
- Ketamine modifies opioid receptor responsivity
- Inhibition of nitric oxide synthase
Ketamine may improve neuropathic pain, in part, by decreasing depression seen in neuropathic pain. The neurological signals for pain do run alongside pathways involved in mood. Emotional pain and physical pain are interrelated. (9)
Ketamine and its antidepressant effects

Although ketamine was discovered in 1962 and FDA approved for anesthesia in 1970, ketamine was quickly recognized as having the potential to augment psychotherapy. the earliest studies were in 1972 in Iran as listed above in the timeline. It wasn’t until the year 2000 when Berman et al. did a small study of ketamine in treatment resistant cases that demonstrated rapid and robust efficacy of ketamine therapy.
At low doses of ketamine, there is a rebalancing of glutamatergic activity and GABA levels due to alterations in intracellular cascades set off by the ketamine therapy. As a simplified level, high levels of glutamate and low GABA levels can lead to anxiety. High levels of glutamate are toxic to neurons. On the other hand, depleted glutamate levels can contribute to depression.
Treatment of depression with ketamine therapy sets off a cascade of interlocking systems. Blocking NMDA receptors at inhibitory interneurons in the brain results in a decrease functioning of GABA neurons that allows for increased activity at specific synapses. The blockage of these interneurons is a ‘go’ signal for the rapid release of glutamate at the synapse, which activates the postsynaptic neuron to produce proteins that allow for increased neural plasticity.
The rapid increase of glutamate in the synapse initially activates AMPA receptors, which results in a calcium influx that depolarizes the neuron. There is a resulting cascade of metabolism that results in the stimulation and formation of brain derived neurotrophic factor (BDNF) and the mTOR pathways. This results in increased synapse formation, increased synaptic strength, and alterations in neurotransmitter control, especially of serotonin and dopamine. The promotion of synaptic growth can “reset” the stress that has caused structural changes in depression. This allows for improved emotional processing as well as improved functioning of memory areas the prefrontal cortex ( executive functioning) and the hippocampus (short-term memory).
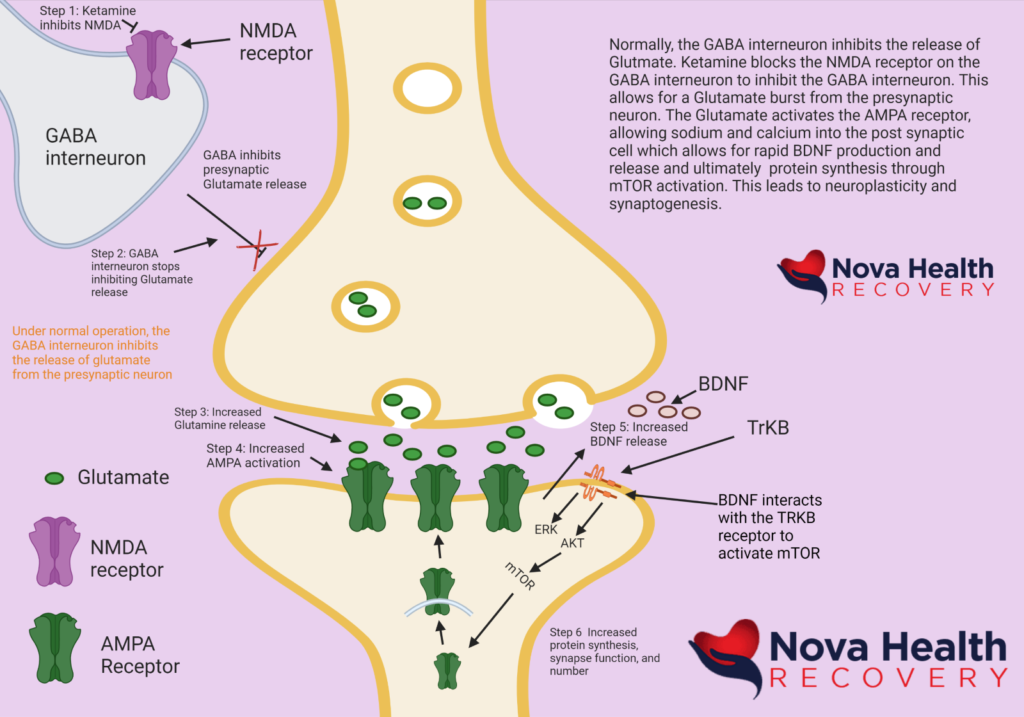
What makes ketamine therapy unique is that it is affecting the glutamatergic system. Looking at the diagram above, in review, the following sequence of events occurs:
- Ketamine binds to the NMDA receptor on the GABA interneuron. This blocks the NMDA receptor and turns off the GABA interneuron.
- The GABA interneuron stops inhibiting glutamate release at the synapse. This is a “go” signal for the release of glutamate from the presynaptic neuron.
- Glutamate is released into the synapse.
- Glutamate interacts with AMPA receptors, causing them to open and allow in sodium, thereby depolarizing the postsynaptic neuron. Generally, excess glutamate is taken up by astrocytes that are part of the synapse. The excess glutamate is converted to glutamine in the astrocytes and then sent back to the presynaptic neuron where it is converted back to glutamate to be reused again. Glutamate can be converted into GABA as well.
- BDNF is released from vesicles and interacts with tyrosine kinase B (TrKB) which then sets off more intracellular cascades to activate mTOR, which results in the production of proteins leading to synapse formation.
What are the risks of ketamine use?
Ketamine is one of the safest drugs available because of its ability to maintain cardiovascular status with minimal support. The World Health Organization has had ketamine on its essential medicines list since 1985. Most cases of death associated with ketamine use are in conjunction with either significant medical conditions or other drug use. Ketamine can certainly be misused and abused, but it has no physiological withdrawal. There could certainly be some psychological dependence, however, if ketamine therapy has effectively made a person less depressed, depression may return after ketamine therapies are stopped. It is of note that when ketamine is used as an antidepressant, the antidepressant effects last way beyond the presence of ketamine in the system. It is not the psychedelic effect of ketamine that is producing the antidepressant effect, but rather the neural plasticity that follows.
Ketamine produces different responses in patients who are depressed versus individuals who are not depressed. Most abuse of ketamine occurs in the context of other drug use in individuals who have no depression, but rather use ketamine for pleasure. One must be careful in the context of the fentanyl epidemic not to accidentally ingest fake ketamine and end up with an overdose of fentanyl. The other issue with snorting ketamine off the street is that it is generally higher doses and anesthetic in nature. Anesthetic doses are not antidepressants as they affect different systems in the brain.
In monitored settings, ketamine is extremely safe, especially in the treatment of mood disorders where the doses are less than 1/10 of what would be used for anesthetic purposes.
There can be changes in heart rate, with an increase in heart rate, as well as blood pressure increases transiently during treatment with ketamine therapy. Other short-term side effects during ketamine therapy include nausea, vomiting, nystagmus, loss of balance, altered states of consciousness, confusion, and sedation.
Ultra-high doses of ketamine that are used to poison rats in controlled studies and in study cases of ketamine abuse as seen in Southeast Asia at grams per day over many months demonstrate the possibility for bladder issues, stomach pain, and alterations of mood. This type of abuse is rarely seen in the United States.
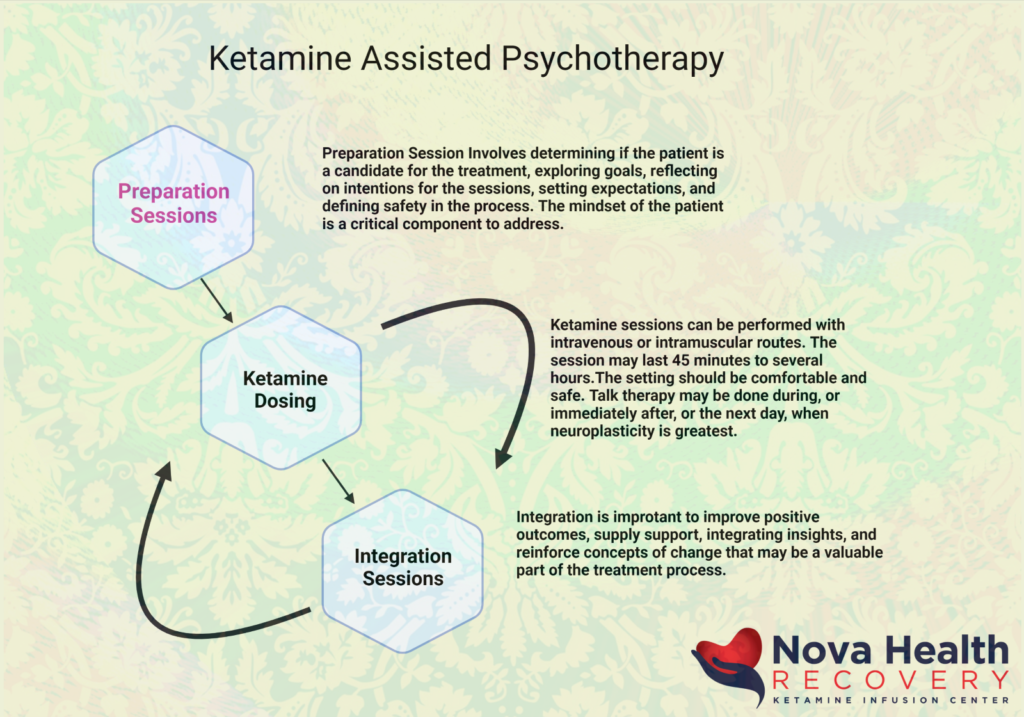
Ketamine Assisted Psychotherapy
Ketamine has been used in the treatment of mood disorders with great efficacy and robust effects. 70% of patients with treatment resistant depression may have significant improvement or remission by the third infusion out of six that are generally given over two weeks. The combination of ketamine therapy with psychotherapy provides a stronger construct for the maintenance of effect with ketamine treatment. The evidence-based medicine behind KAP is explored in another article, but generally there are preparatory sessions, in which the person’s eligibility for treatment is defined and establishing treatment goals and expectations. The person then receives, at a later time, the ketamine dosing, whether it is by intravenous or intramuscular approaches, with sessions lasting between 45 minutes to several hours in length. Psychotherapy sessions can be applied during the ketamine experience or immediately after as an integration. Subsequent integration therapy is critical when the person has returned from their altered states of consciousness in order to focus on positive outcomes, supply support, integrating insights, and reinforcing concepts of change that may be a valuable part of the treatment process.
1 Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018 Apr;23(4):801-811. doi: 10.1038/mp.2017.255. Epub 2018 Mar 13. PMID: 29532791; PMCID: PMC5999402.
2. Muller J, Pentyala S, Dilger J, Pentyala S. Ketamine enantiomers in the rapid and sustained antidepressant effects. Ther Adv Psychopharmacol. 2016 Jun;6(3):185-92. doi: 10.1177/2045125316631267. Epub 2016 Mar 10. PMID: 27354907; PMCID: PMC4910398.
3 Jelen LA, Young AH, Stone JM. Ketamine: A tale of two enantiomers. J Psychopharmacol. 2021 Feb;35(2):109-123. doi: 10.1177/0269881120959644. Epub 2020 Nov 6. PMID: 33155503; PMCID: PMC7859674.
4 Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, Dong C, Hashimoto K. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015 Sep 1;5(9):e632. doi: 10.1038/tp.2015.136. Erratum in: Transl Psychiatry. 2020 Aug 21;10(1):295. PMID: 26327690; PMCID: PMC5068814.
5 Zhang JC, Li SX, Hashimoto K. R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacol Biochem Behav. 2014 Jan;116:137-41. doi: 10.1016/j.pbb.2013.11.033. Epub 2013 Dec 3. PMID: 24316345.
6 Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KS, Fang Y, Huang XP, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA Jr, Gould TD. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016 May 26;533(7604):481-6. doi: 10.1038/nature17998. Epub 2016 May 4. PMID: 27144355; PMCID: PMC4922311.
7 Hashimoto K. Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem Pharmacol. 2020 Jul;177:113935. doi: 10.1016/j.bcp.2020.113935. Epub 2020 Mar 26. PMID: 32224141.
8 Sarton E, Teppema LJ, Olievier C, Nieuwenhuijs D, Matthes HW, Kieffer BL, Dahan A. The involvement of the mu-opioid receptor in ketamine-induced respiratory depression and antinociception. Anesth Analg. 2001 Dec;93(6):1495-500, table of contents. doi: 10.1097/00000539-200112000-00031. PMID: 11726430.
9. Wang J, Goffer Y, Xu D, Tukey DS, Shamir DB, Eberle SE, Zou AH, Blanck TJ, Ziff EB. A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology. 2011 Oct;115(4):812-21. doi: 10.1097/ALN.0b013e31822f16ae. PMID: 21934410; PMCID: PMC3222930.